This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
What is phylogeny?
Phylogenetics is the study of evolutionary relationships between organisms to determine relative relatedness. A common way to study phylogenetics was to compare morphology between organisms and use this information to determine relatedness. For example, using morphological based techniques, bats and birds would be closely related because their wings have similar functions; however, the wings on these two organisms are very different structurally. Molecular phylogeny compares the relatedness between organisms by analyzing the differences between specific gene sequences.[1] This can be more specific than morphological approaches, and if used in conservative sequences it can be very specific and aide in distinguishing between species. A common way to visualize relatedness is by creating a phylogenetic tree, which links organisms based on their last common ancestor.
Molecular Phylogenetics of TMC6
By using molecular phylogenetic techniques the TMC6 gene can be analyzed and compared between species directly, giving a more specific comparison between organisms. Assembling and generating the tree commonly use one of three different methods: Maximum likelihood, Neighbor joining, or Average distance.
Neighbor Joining: Uses the similarity scores between the organisms that are generated by the percent similarity to generate the initial tree. It then calculates the branch length by determining the percent change that occurred since the last common ancestor. [2]
Average Distance: Uses the similarity scores to generate the tree, but then assumes that each species diverges the same amount from the last common ancestor and draws the tree as such. [2]
Maximum Likelihood: Initially a tree is built using one of the above methods, but then the branch lengths are adjusted to maximize the likelihood of the data set for that tree topology. [2]
Neighbor Joining: Uses the similarity scores between the organisms that are generated by the percent similarity to generate the initial tree. It then calculates the branch length by determining the percent change that occurred since the last common ancestor. [2]
Average Distance: Uses the similarity scores to generate the tree, but then assumes that each species diverges the same amount from the last common ancestor and draws the tree as such. [2]
Maximum Likelihood: Initially a tree is built using one of the above methods, but then the branch lengths are adjusted to maximize the likelihood of the data set for that tree topology. [2]
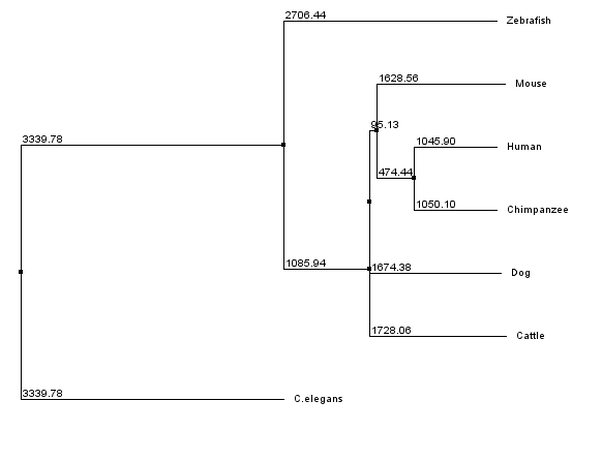
Conclusion: By using the neighbor joining method to analyze the differences between the TMC6 gene and generate the tree it was shown that the most closely related homolog to humans is in Chimpanzees. The second closest homolog to humans is in mice, and the least related homolog is found in C. elegans. This information can be used to predict how similar the genes will function compared to the human TMC6 gene.
Sources:
1. https://www.encyclopedia.com/earth-and-environment/ecology-and-environmentalism/environmental-studies/phylogenetics
2. https://genetics564.weebly.com/homology--phylogeny.html
1. https://www.encyclopedia.com/earth-and-environment/ecology-and-environmentalism/environmental-studies/phylogenetics
2. https://genetics564.weebly.com/homology--phylogeny.html
